


Medical device regulation
Myron Yaster MD and Mark Schreiner MD

Virtually everything we use when we provide an anesthetic – drugs or medical devices and equipment – is regulated in some fashion by governmental agencies like the U.S. Food and Drug Administration (FDA) or the European Medicines Agency (EMA). These regulations help ensure the public safety and the efficacy of the products we use. I think most understand the long, arduous, and very expensive path required for new drug development. But what about medical devices? Just think about the arms race amongst your anesthesia machine manufacturers: ventilators that mimic ICU ventilators, built in checklists to perform when a machine is turned on; integration with electronic medical records, fail safe designs etc. But, were any of these “advances” and new machines actually tested as part of clinical trials before they were released for clinical use? Or think about IV or peripheral nerve block catheters. How were the materials and new designs tested or were they ever tested for safety in animals or human trials before they were introduced into our practices? Unfortunately, the answer is rarely.
In today’s PAAD we will review 2 articles by Kadakia et al.1,2 that discuss how the regulation and testing of medical devices. I think many of you will be very surprised by these articles. But first an important word of disclosure. Over the course of my career, I was heavily involved with several medical device companies, particularly Cook Medical (https://www.cookmedical.com/ ) and Smiths medical (now ICU Medical: (https://www.icumed.com/) in the design of several products that all of you use on a daily basis, including central line and peripheral and central neural blockade catheter design and packaging, epidural needle design, IVPCA device design and programming, and infusion pump technologies to name just a few. Indeed, some of this consulting started almost at the beginning of my career when working with Cook and its product engineers, I helped in the transition from polyethylene central line catheters to polyurethane and silicone catheters. To be honest, I loved working with Cook and Smiths because they, unlike many other companies, were always interested in developing products and design solutions for the pediatric population. Some of the very best and most ethical people I ever dealt with during my career worked for these companies. (I think this could be shortened. It is longer than necessary.)
As I said in my opening comments, today’s PAAD may be an eye opener for many of you. I’ve asked my good friend and colleague, Dr. Mark Schreiner who during his career was actively involved in new drug development and product labeling to help. Finally, these were issues that were very important to the late Dr. Ron Litman who started the PAAD and in whose memory we continue to publish it. Myron Yaster MD
Original article
Kadakia KT, Rathi VK, Dhruva SS, Ross JS, Krumholz HM. Modernizing Medical Device Regulation: Challenges and Opportunities for the 510(k) Clearance Process. Ann Intern Med. 2024 Nov;177(11):1558-1565. doi: 10.7326/ANNALS-24-00728. Epub 2024 Oct 8. PMID: 39374526.
Original article
Kadakia KT, Bikdeli B, Gupta A, Dhruva SS, Ross JS, Krumholz HM. Information Disclosure, Medical Device Regulation, and Device Safety: The Case of Cook Celect IVC Filters. Ann Intern Med. 2024 Nov 19. doi: 10.7326/ANNALS-24-00089. Epub ahead of print. PMID: 39556835.
Why are medical devices treated differently by the FDA than new drugs? Surprisingly, until 1976 devices weren’t regulated at all. Congress passed the Medical Device Amendments of 1976 to the Federal Food, Drug, and Cosmetic Act which classified devices as low, moderate or high risk. Only those that are high risk usually require clinical premarket testing. Almost all devices that are moderate risk are approved via the 510(k) pathway provided for in the 1976 act. The basic idea of the 510(k) pathway is that if there is an existing marketed medical device (a predicate), then minor modifications to that device would not generally require clinical testing if the new device was substantially equivalent (not initially defined). The key issue is defining what is substantially equivalent.
Subsequently, Kadakia notes “Nearly all medical devices reviewed by the U.S. Food and Drug Administration (FDA) are authorized via the 510(k) clearance process.” “This evaluation usually does not require clinical evidence of safety and effectiveness. Advocates of the 510(k) clearance process tout its support for device innovation and rapid market access, and critics of the 510(k) clearance process express that it may inadequately protect patient safety. In September 2023, the FDA issued 3 guidance documents that, if finalized, would significantly change medical device regulation. This article provides clinical and regulatory context for the proposed guidance documents, which focus on predicate selection, clinical testing requirements, and implantable devices, and identifies opportunities for further reforms that promote transparency and patient safety.”1
The 510(k) approval process has been heavily criticized with the IOM recommending elimination entirely. Instead, the FDA has made efforts to reform the process. Rathi et al. succinctly summarized the main issues as follows: “… there are several reasons why the 510(k) pathway may permit clearance of devices that have undergone insufficient testing1: a reliance on lenient standards for “substantial equivalence,” which facilitates clearance of poorly understood technologies; device “creep,” whereby iterative design changes result in unproven devices dissimilar from original predicates; and the use of unsafe or outdated predicates.”7
Over time drastic changes evolve the new device so that it can be far afield from the original, clinically tested device. This has resulted in approval of devices with disasterous consequences. The figure from Ardaugh et al.8 shows the complex ancestral tree that led to metal on metal hip joint replacements. The small iterations accumulated until the devices were far afield from the originally tested device and include several discontinued devices.
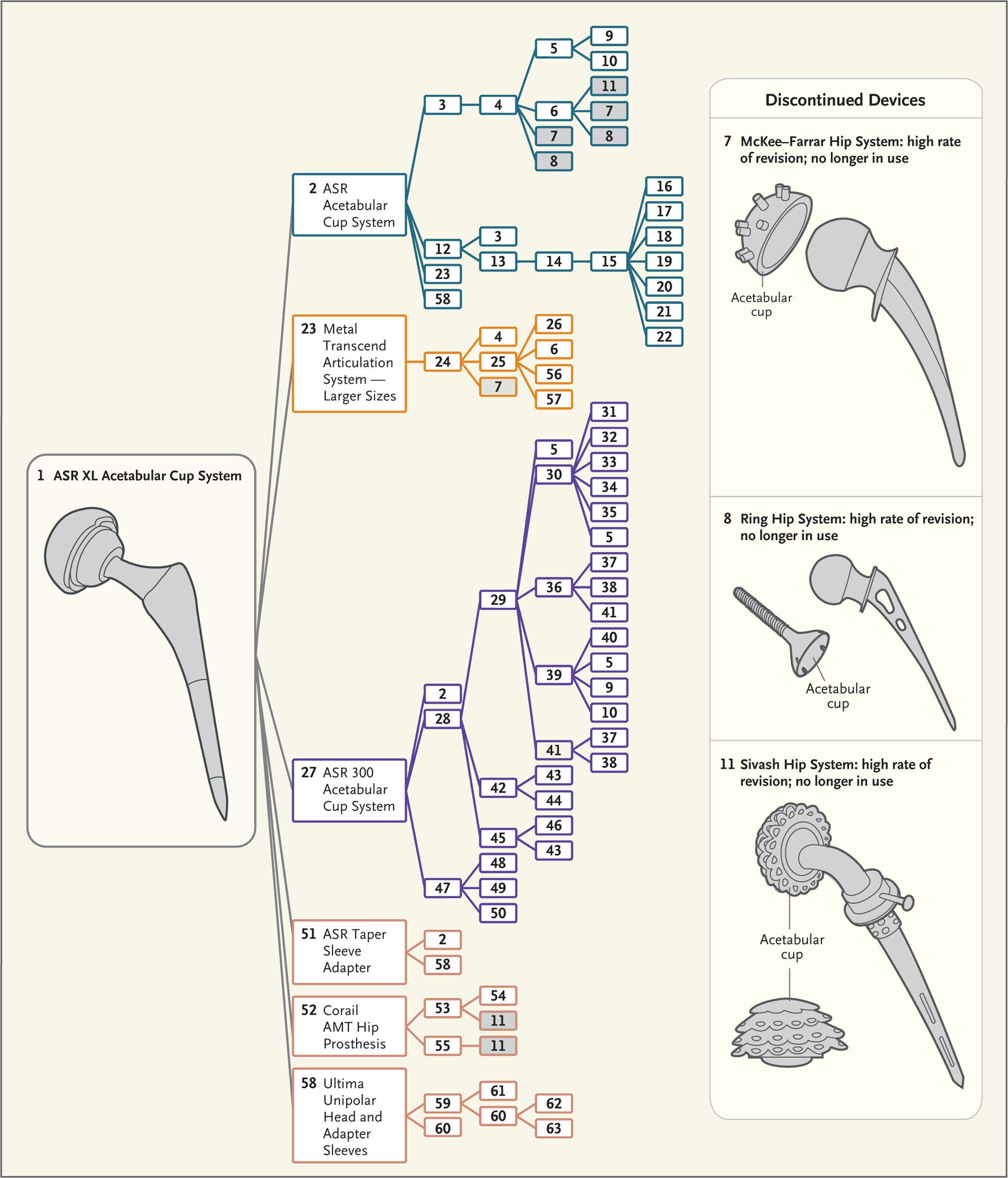
Each number represents a corresponding number in the Supplementary Appendix (available at NEJM.org), where the device names, companies, 510(k) numbers, and decision dates for most devices can be found.
Multiple studies have estimated that the research and development (R&D) cost for a new drug ranges from $314 million to $4.46 billion, depending on the therapeutic area, data, and modeling assumptions.3,4 This unbelievably high cost of drug development not only drives the astronomical cost of the drugs we use in medical practice but hampers innovation and limits new drug entry into the marketplace. In return, the public is ensured of the safety and efficacy of new drugs.
The 510(k) process is much simpler and cheaper. The FDA authorizes approximately 3000 new devices annually.5 Think of how expensive it would be if devices had to go through the same process as medications. As an example, consider central line catheters which often introduce a new material such as polyurethane for polyethylene or an antibiotic coating or a change the length of the catheter. Provided there is non-clinical (animal) data or another predicate device supporting the use of the new material these changes would be considered minor modifications of an approved product already in clinical use. Animal testing and bench testing can avoid the need for clinical trials. Thus, the 510(k) process supports innovation without compromising safety,6 and is the regulatory process, through which 99% of devices (primarily moderate risk) are authorized. Most importantly, “unlike drug approvals, obtaining 510(k) clearance is not a test of safety and efficacy, but rather a demonstration that new devices are similar to already authorized devices.”1
What else could go wrong? Unfortunately, as can be seen in their case study of the Cook Celect IVC filters2quite a lot! “Recently unsealed court documents from litigation related to Celect reveal that the device’s clinical study protocol did not follow FDA guidance for IVC filter testing and that study outcome definitions for IVC perforation had lower sensitivity for detecting adverse events than those recommended by professional societies. Furthermore, a comparison of court documents and the public record indicates that adverse events and patient deaths were misreported to FDA reviewers and were inaccurately reported in the published literature and on the device label, providing clinicians and the public with inaccurate information about the device’s safety. The Celect IVC filter case demonstrates the need for regulatory reforms to ensure that critical safety data are accessible to the FDA, clinicians, and patients to inform decision making.”2
Is this an isolated story or becoming an industry norm? As I (MY) stated in the introduction, I’ve worked extensively with the folks at Cook over several decades and always thought of them as the best of the best in terms of innovation, product design, and commitment to patient safety. If this can happen at Cook it can happen to any company that develops and markets medical devices. New regulations and transparency are clearly required. As highlighted by Katakia, the FDA issued 3 new draft guidance documents in September 2023 to address issues with the 510(k) pathway. What will actually happen remains to be seen.
What do you think? Send your thoughts and comments to Myron who will post in a Friday reader response.
References
1. Kadakia KT, Rathi VK, Dhruva SS, Ross JS, Krumholz HM. Modernizing Medical Device Regulation: Challenges and Opportunities for the 510(k) Clearance Process. Annals of internal medicine 2024;177(11):1558-1565. (In eng). DOI: 10.7326/annals-24-00728.
2. Kadakia KT, Bikdeli B, Gupta A, Dhruva SS, Ross JS, Krumholz HM. Information Disclosure, Medical Device Regulation, and Device Safety: The Case of Cook Celect IVC Filters. Annals of internal medicine 2024 (In eng). DOI: 10.7326/annals-24-00089.
3. Sertkaya A, Beleche T, Jessup A, Sommers BD. Costs of Drug Development and Research and Development Intensity in the US, 2000-2018. JAMA network open 2024;7(6):e2415445-e2415445. DOI: 10.1001/jamanetworkopen.2024.15445.
4. Wouters OJ, McKee M, Luyten J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. Jama 2020;323(9):844-853. (In eng). DOI: 10.1001/jama.2020.1166.
5. Darrow JJ, Avorn J, Kesselheim AS. FDA Regulation and Approval of Medical Devices: 1976-2020. Jama 2021;326(5):420-432. (In eng). DOI: 10.1001/jama.2021.11171.
6. Dhruva SS, Kesselheim AS, Woloshin S, et al. Physicians' Perspectives On FDA Regulation Of Drugs And Medical Devices: A National Survey. Health affairs (Project Hope) 2024;43(1):27-35. (In eng). DOI: 10.1377/hlthaff.2023.00466.
7. Rathi VK, Ross JS. Modernizing the FDA's 510(k) Pathway. N Engl J Med. 2019 Nov 14;381(20):1891-1893. PMID: 31644865.
8. Ardaugh BM, Graves SE, Redberg RF. The 510(k) ancestry of a metal-on-metal hip implant. N Engl J Med. 2013 Jan 10;368(2):97-100. PMID: 23301729.