


CF is no longer Cystic Fibrosis but rather Cure Found!
Myron Yaster MD

In a world that increasingly appears to have gone mad, it is easy to overlook miracles. And what is happening in cystic fibrosis is nothing short of a medical miracle. When I was starting out as a first year resident in Pediatrics in the 1970s, most children born with CF died in the first decade of life. Today, life expectancy extends into the 5th and 6th decades and beyond. Today’s PAAD by Grasemann and Ratjen1 in the New England J of Medicine reviews the pathophysiology, evaluation, and treatment of cystic fibrosis, including recent advances with the use of highly effective modulator therapy. Although we see fewer and fewer of these patients in the OR, and when we do they are usually getting older and older, the unprecedented improvement in the health of these “pediatric” patients is so monumental that I thought this review would be ideally suited for the PAAD readership. Myron Yaster MD
Original article
Grasemann H, Ratjen F. Cystic Fibrosis. N Engl J Med. 2023 Nov 2;389(18):1693-1707. doi: 10.1056/NEJMra2216474. PMID: 37913507.
“Cystic fibrosis is an autosomal recessive disease caused by variants in the CFTR gene. CFTR encodes for an ion channel, CFTR, that is involved in regulation of the water–electrolyte balance on the surface of many organ systems, including the upper and lower airways, intestine, pancreas, biliary tree, cervix, vas deferens, and sweat glands.2”1 Although there are almost 700 disease causing variants, “deletion of three base pairs in CFTR leading to the loss of the amino acid phenylalanine at position 508 (F508del) of the protein is the most common (85%) cystic fibrosis–causing variant.”1
“Cystic fibrosis–causing variants have different implications with respect to CFTR production, processing, expression, and function. They can result in premature termination codons with reduced or absent CFTR formation (class I variant); premature degradation due to protein misfolding (class II); abnormalities of channel gating (class III), conductance (class IV), or both; reduced transcript, promotor, or splicing abnormalities (class V); or accelerated turnover from the cell surface (class VI).”1 A given variant may have more than one molecular defect and all are the targets of drug therapy (figure 1). The manifestations of CF on major organs and on symptoms are seen in figure 2.
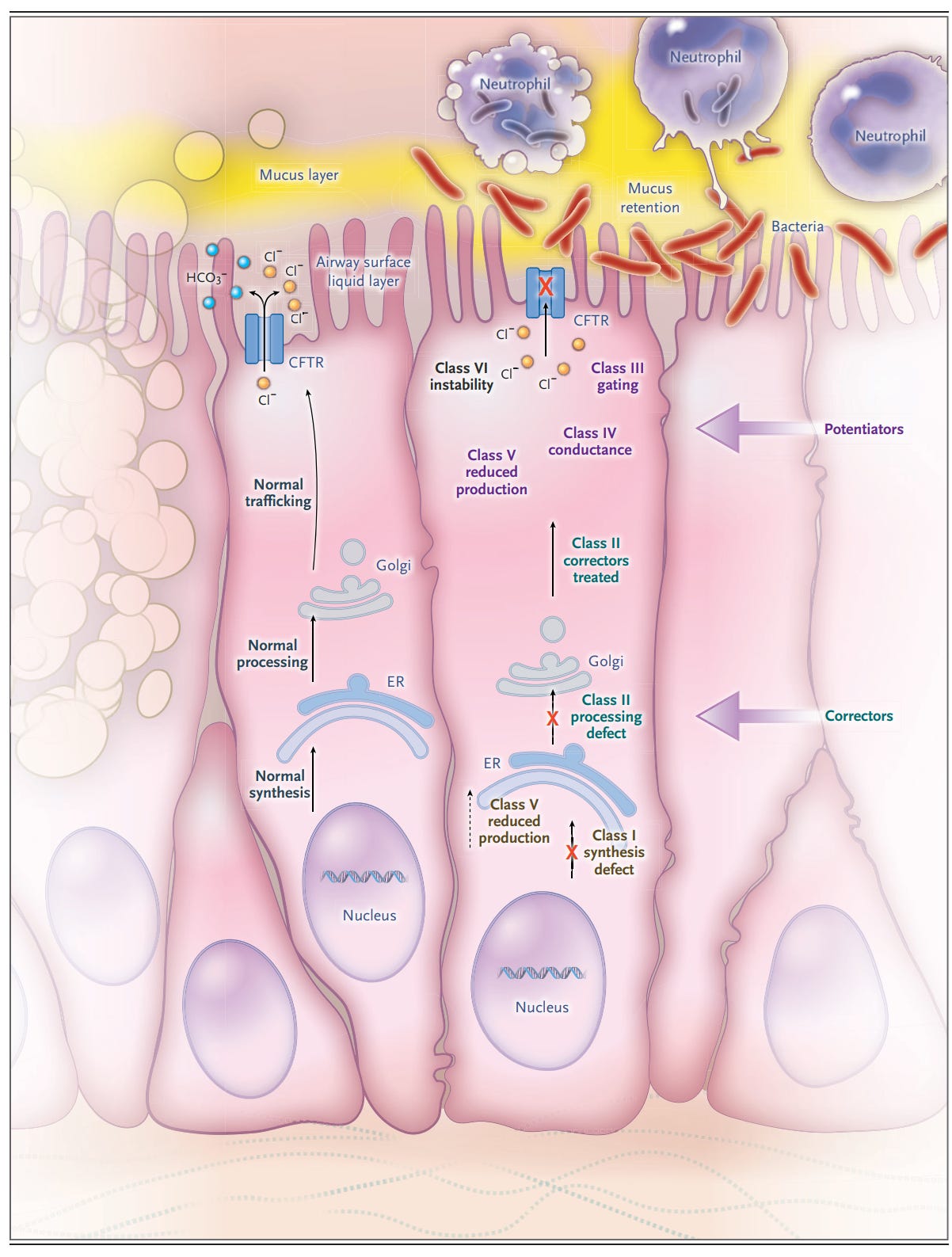

We think for many of you, the clinical manifestations of CF and how the disease is diagnosed are known. What is new, really miraculous is the revolution in treatment.
Treatment: CF modulators: Take a close look at figure 1. Correctors improve trafficking and processing of CFTR protein to the apical membrane of the cell. Potentiators improve function of CFTR expressed at the cell membrane. The potentiator ivacaftor is effective in persons with cystic fibrosis carrying variants with residual CFTR expression in which channel conductance is reduced. “Ivacaftor binds to CFTR and is thought to thereby increase the probability of opening the channel and has led to unprecedented improvements in respiratory outcomes, including lung function, the risk of pulmonary exacerbation, and respiratory symptoms as well as nutritional status and other manifestations of cystic fibrosis.”1 Because only a small number (5%) of CF patients respond to only ivacaftor, “combination therapy with corrector drugs that ameliorate the protein-folding defect (elexacaftor–tezacaftor) and allow for transport of the channel to the cell surface and ivacaftor to improve channel gating are warranted to achieve clinical benefit.”1 This triple therapy has improved lung function exceeding the previously observed results with ivacaftor alone in gating variants.3,4 Depending on the variant, not all patients respond and the use of triple therapy. Further, until recently, the use of these drugs was limited to older children. However, this is rapidly changing. In fact, triple therapy is being studied in younger and younger children with rather dramatic effect. Indeed, it is suspected that starting this therapy in the very young (< 1 year of age) may prevent other systemic manifestations of CF like diabetes!
It is beyond the scope of the PAAD to discuss symptom management of patients with CF like lung infection, acute pulmonary exacerbations, diabetes, nutritional and infertility management. I refer you to the article for a deeper dive into this. Further, there is no mention in this article of the anesthetic management of these patients who often present to the OR for diagnostic and therapeutic bronchoscopies and central line and feeding tube placement. For a detailed review of perioperative anesthetic management of CF patients, please see the article by Lee et al.5 which we discussed in a previous PAAD.
On the other hand, in today’s article, Grasemann et al.1 point out that “the CFTR modulators are substrates and inducers of the cytochrome P450 enzyme and interact with other drugs that are metabolized in this pathway.” 1,6 Many of the drugs we use in our anesthesia practice are metabolized in these pathways. Should we adjust or even discontinue the CFTR modulator drugs perioperatively? Are there significant side effects and drug–drug interactions?6 No one knows and I think this would be an ideal future multi-institutional study or registry for SPA researchers to perform in order to monitor adverse events in people with CF being treated with CFTR modulators in a real world perioperative setting.6
I want to leave you with a sense of wonder at the miracle that has occurred before our eyes. The dream that CF would mean Cure Found is here. What are your thoughts? Have you experienced treating patients on triple therapy? Send your comments and thoughts to Myron who will post in a Friday Reader response.
References
1. Grasemann H, Ratjen F: Cystic Fibrosis. N Engl J Med 2023; 389: 1693-1707
2. Stoltz DA, Meyerholz DK, Welsh MJ: Origins of cystic fibrosis lung disease. N Engl J Med 2015; 372: 351-62
3. Sutharsan S, McKone EF, Downey DG, Duckers J, MacGregor G, Tullis E, Van Braeckel E, Wainwright CE, Watson D, Ahluwalia N, Bruinsma BG, Harris C, Lam AP, Lou Y, Moskowitz SM, Tian S, Yuan J, Waltz D, Mall MA: Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: a 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir Med 2022; 10: 267-277
4. Flume PA, Biner RF, Downey DG, Brown C, Jain M, Fischer R, De Boeck K, Sawicki GS, Chang P, Paz-Diaz H, Rubin JL, Yang Y, Hu X, Pasta DJ, Millar SJ, Campbell D, Wang X, Ahluwalia N, Owen CA, Wainwright CE: Long-term safety and efficacy of tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years or older who are homozygous or heterozygous for Phe508del CFTR (EXTEND): an open-label extension study. Lancet Respir Med 2021; 9: 733-746
5. Lee AJ, Huffmyer JL, Thiele EL, Zeitlin PL, Chatterjee D: The Changing Face of Cystic Fibrosis: An Update for Anesthesiologists. Anesth Analg 2022
6. Dagenais RVE, Su VCH, Quon BS: Real-World Safety of CFTR Modulators in the Treatment of Cystic Fibrosis: A Systematic Review. J Clin Med 2020; 10